Computational Protein Design and Modeling
Predicting and designing the structures of proteins with biologically useful accuracy has been a key challenge in computational structural biology and molecular engineering. We have made methodological advances that address one of the main bottlenecks: sampling the vast number of conformations accessible to proteins. We have utilized a method for moving through conformational space inspired by principles from robotics – a field with a rich history in efficient calculation of mechanically accessible states subject to constraints. We applied the same mathematics that can be used to direct the motions of a robot arm to compute the degrees of freedom of a polypeptide chain (Mandell et al., Nature Methods 2009). Our predictions generate hypotheses on protein conformations controlling biological processes – such as protein recognition, signal transduction, and enzyme active site gating – and are laying the foundation for our work reengineering and “reshaping” protein interfaces and active sites for new functions.
|
|
Selected publications
|
|
ROBOTICS-INSPIRED CONFORMATIONAL SAMPLING IN PROTEIN LOOP RECONSTRUCTION
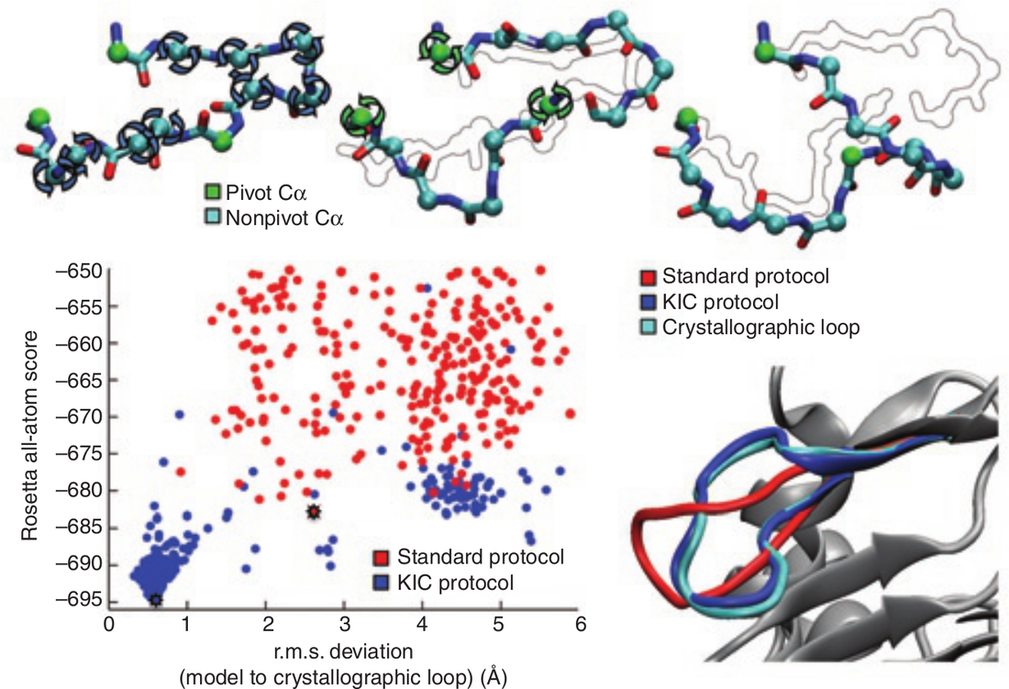
Sub-Angstrom Accuracy In Protein Loop Reconstruction By Robotics-Inspired Conformational Sampling
Mandell, DJ, Coutsias, EA, Kortemme, T Nat Methods 6(8):551-2, 2009 |
Proteins demonstrate considerable conformational diversity that facilitates both recognition of cognate biomolecules as well as accommodation of amino acid sequence mutations that confer new functions. Our ability to engineer proteins with new capabilities depends directly on the accuracy to which we can predict conformational changes in response to binding or mutagenesis.
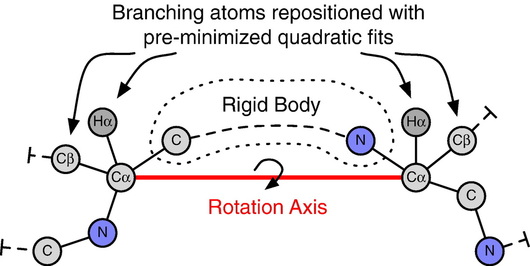
In this work we developed a method for rapidly sampling polypeptide conformations called kinematic closure (KIC), which is based on tools and methods used in the field of robotics. In this formulation, KIC analytically determines all possible values for 6 backbone torsions of a polypeptide segment while efficiently sampling any remaining degrees of freedom, including other torsions, bond angles, and bond lengths. Since the solutions are exact, changes to the polypeptide are local—they do not propagate beyond the sampled segment. We implemented KIC within Rosetta, and then used it to achieve unprecedented sub-Angstrom median accuracy in predicting the crystallographic conformations of a well-studied dataset of 12-residue protein loops, which are typically the most difficult regions to model due to their lack of secondary structure. |
Subsequently, we extended KIC through detailed enhancements to the sampling and scoring methods that enable median sub-Angstrom reconstruction of loops 14-17 residues in length, a regime of that was previously inaccessible to high-resolution prediction due to combinatoric explosion of possible conformations. Further, by iterating KIC moves throughout a protein backbone we have created whole-protein structural ensembles for flexible backbone sequence design that recapitulate 70% of the sequences observed in comprehensive phage display experiments, a figure that compares favorably to alternative methods for local or global backbone sampling. Together, these results indicate KIC provides a highly-accurate move for sampling protein loop conformations, as well as sampling protein backbone conformational ensembles.
BACKRUB-LIKE BACKBONE SIMULATION TO RECAPITULATE NATURAL PROTEIN CONFORMATIONAL VARIABILITY
Backrub-Like Backbone Simulation Recapitulates Natural Protein Conformational Variability And Improves Mutant Side-Chain Prediction
Smith, CA, Kortemme, T J Mol Biol 380(4):742-56, 2008 |
Proteins undergo conformational fluctuations in response to thermal energy, binding events, and mutation. Understanding and predicting such excursions around the native state of a protein is a key challenge in computational molecular biology. However, many applications keep the backbone structure fixed, while in actual proteins the backbone often undergoes subtle shifts in response to binding events or sequence changes. Successfully capturing such near-native shifts is thus important for many docking and design applications.
In this work, we developed a new local perturbation of protein backbones based on motions seen in high-resolution crystal structures. These fluctuations observed in the crystal lattice motivated Davis et al. to create a simple model, called “Backrub”, for subtle backbone shifts using just three residues. The core idea the work here is to use that type of motion, observed in nature, to computationally sample backbone configurations in a generalized scheme. |
We implemented a backrub-inspired sampling method in the Rosetta structure prediction and design program. We evaluated this model of backbone flexibility using three different tests: First, we show that Rosetta backrub simulations recapitulate the correlation between backbone and side-chain conformations in the high-resolution crystal structures upon which the model was based. Next, we show that backbone flexibility improves the accuracy of predicting point-mutant side-chain conformations over fixed backbone rotameric sampling alone. Finally, we show that backrub sampling of triosephosphate isomerase loop 6 can capture the millisecond/microsecond oscillation between the open and closed states observed in solution. Our results suggest that backrub sampling captures a sizable fraction of localized conformational changes that occur in natural proteins.