SUB-ANGSTROM ACCURACY IN PROTEIN LOOP RECONSTRUCTION
BY ROBOTICS-INSPIRED CONFORMATIONAL SAMPLING
BY ROBOTICS-INSPIRED CONFORMATIONAL SAMPLING
Sub-Angstrom Accuracy In Protein Loop Reconstruction By Robotics-Inspired Conformational Sampling
Mandell, DJ, Coutsias, EA, Kortemme, T
Nat Methods 6(8):551-2, 2009
Mandell, DJ, Coutsias, EA, Kortemme, T
Nat Methods 6(8):551-2, 2009
Proteins demonstrate considerable conformational diversity that facilitates both recognition of cognate biomolecules as well as accommodation of amino acid sequence mutations that confer new functions. Our ability to engineer proteins with new capabilities depends directly on the accuracy to which we can predict conformational changes in response to binding or mutagenesis.
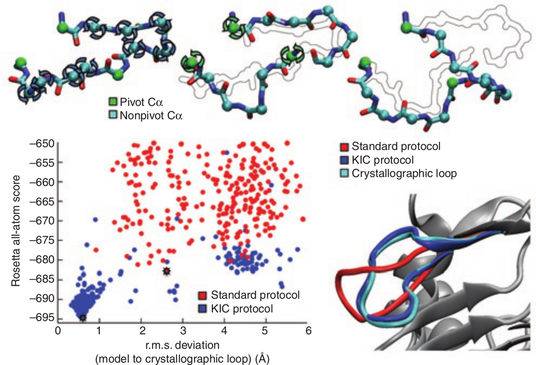
In this work we developed a method for rapidly sampling polypeptide conformations called kinematic closure (KIC), which is based on tools and methods used in the field of robotics. In this formulation, KIC analytically determines all possible values for 6 backbone torsions of a polypeptide segment while efficiently sampling any remaining degrees of freedom, including other torsions, bond angles, and bond lengths. Since the solutions are exact, changes to the polypeptide are local—they do not propagate beyond the sampled segment.
We implemented KIC within Rosetta, and then used it to achieve unprecedented sub-Angstrom median accuracy in predicting the crystallographic conformations of a well-studied dataset of 12-residue protein loops, which are typically the most difficult regions to model due to their lack of secondary structure. Subsequently, we extended KIC through detailed enhancements to the sampling and scoring methods that enable median sub-Angstrom reconstruction of loops 14-17 residues in length, a regime of that was previously inaccessible to high-resolution prediction due to combinatoric explosion of possible conformations. Further, by iterating KIC moves throughout a protein backbone we have created whole-protein structural ensembles for flexible backbone sequence design that recapitulate 70% of the sequences observed in comprehensive phage display experiments, a figure that compares favorably to alternative methods for local or global backbone sampling. Together, these results indicate KIC provides a highly-accurate move for sampling protein loop conformations, as well as sampling protein backbone conformational ensembles.
In this work we developed a method for rapidly sampling polypeptide conformations called kinematic closure (KIC), which is based on tools and methods used in the field of robotics. In this formulation, KIC analytically determines all possible values for 6 backbone torsions of a polypeptide segment while efficiently sampling any remaining degrees of freedom, including other torsions, bond angles, and bond lengths. Since the solutions are exact, changes to the polypeptide are local—they do not propagate beyond the sampled segment.
We implemented KIC within Rosetta, and then used it to achieve unprecedented sub-Angstrom median accuracy in predicting the crystallographic conformations of a well-studied dataset of 12-residue protein loops, which are typically the most difficult regions to model due to their lack of secondary structure. Subsequently, we extended KIC through detailed enhancements to the sampling and scoring methods that enable median sub-Angstrom reconstruction of loops 14-17 residues in length, a regime of that was previously inaccessible to high-resolution prediction due to combinatoric explosion of possible conformations. Further, by iterating KIC moves throughout a protein backbone we have created whole-protein structural ensembles for flexible backbone sequence design that recapitulate 70% of the sequences observed in comprehensive phage display experiments, a figure that compares favorably to alternative methods for local or global backbone sampling. Together, these results indicate KIC provides a highly-accurate move for sampling protein loop conformations, as well as sampling protein backbone conformational ensembles.